
Publications
Publications
View all publications from the Jänne Lab.
featured publications
Seshiru Nakazawa, MD, PhD, Federica Pecci, MD, Biagio Ricciuti, MD, PhD, Felix H. Gottlieb, BS, Francesco Facchinetti, MD, PhD, Guilherme Harada, MD, Monica F. Chen, MD, Matteo Repetto, MD, Flavia Giacomini, MSc, Jie Jiang, PhD, Marie-Anaïs Locquet, PhD, Maisam Makarem, MD, PhD, Joao V. Alessi, MD, Alessandro Di Federico, MD, Mihaela Aldea, MD, PhD, Edoardo Garbo, MD, Malini M. Gandhi, MD, Arushi Saini, Danielle Haradon, Magda Bahcall, PhD, William W. Feng, PhD, Jessica K. Lee, PhD, Alexa B. Schrock, PhD, Alexander Drilon, MD, Mark M. Awad, MD, PhD, Pasi A. Jänne, MD, PhD
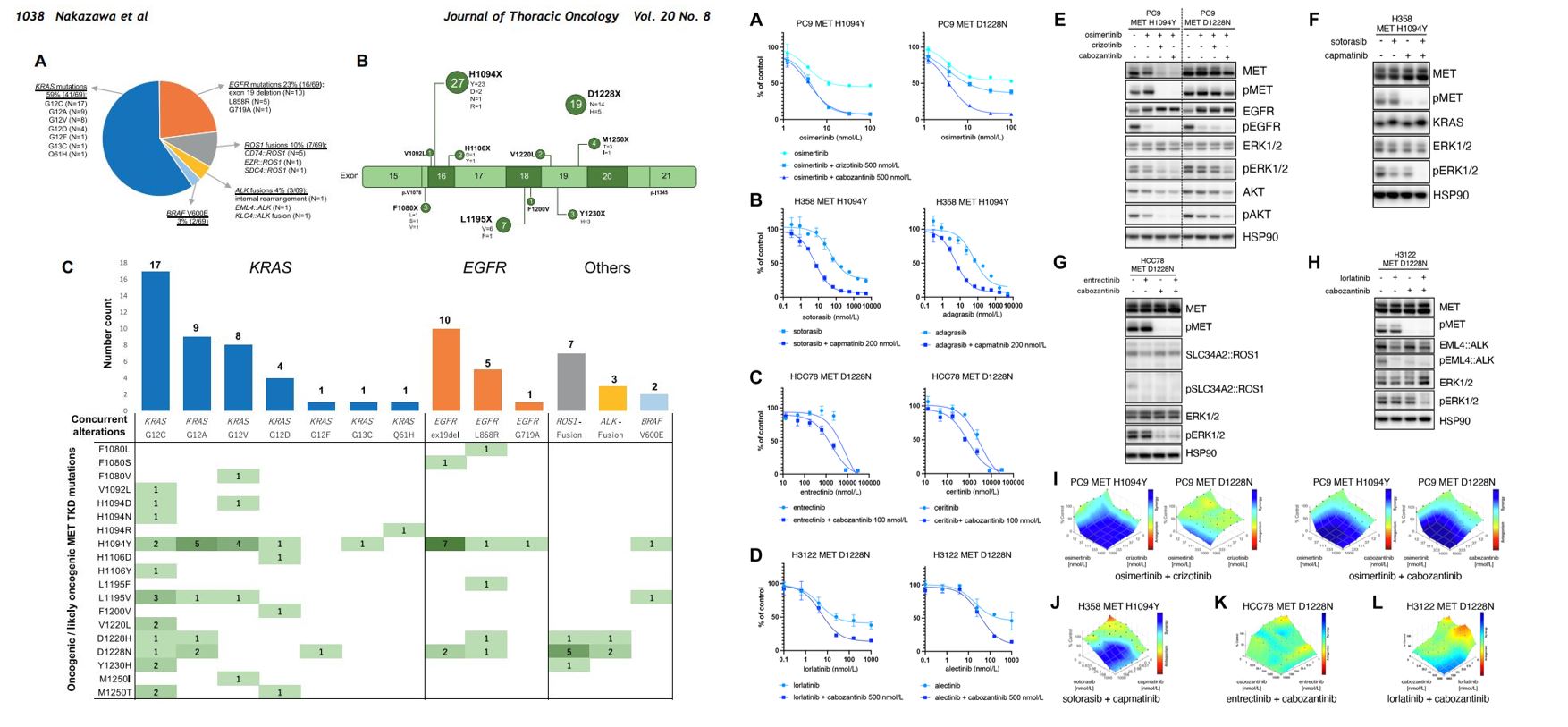
Activating Mutations in the MET Kinase Domain Co-Occur With Other Driver Oncogenes and Mediate Resistance to Targeted Therapy in NSCLC
Abstract:
Allosteric inhibition of drug resistant forms of EGFR L858R mutant NSCLC
Ciric To, Tyler S. Beyett, Jaebong Jang, William W. Feng, Magda Bahcall, Heidi M. Haikala, Bo H. Shin, David E. Heppner, Jaimin K. Rana, Brittaney A. Leeper, Kara M. Soroko, Michael J. Poitras, Prafulla C. Gokhale, Yoshihisa Kobayashi, Kamal Wahid, Kari J. Kurppa , Thomas W. Gero, Michael D. Cameron, Atsuko Ogino, Mierzhati Mushajiang, Chunxiao Xu, Yanxi Zhang, David A. Scott4 , Michael J. Eck, Nathanael S. Gray, Pasi A. Jänne.

Allosteric inhibitor JBJ-09-063 effectively treats drug resistant forms of EGFR L858R mutant non-small cell lung cancer
Abstract:
Epidermal growth factor receptor (EGFR) therapy using small-molecule tyrosine kinase inhibitors (TKIs) is initially efficacious in patients with EGFR-mutant lung cancer, although drug resistance eventually develops. Allosteric EGFR inhibitors, which bind to a different EGFR site than existing ATP-competitive EGFR TKIs, have been developed as a strategy to overcome therapy-resistant EGFR mutations. Here we identify and characterize JBJ-09-063, a mutant-selective allosteric EGFR inhibitor that is effective across EGFR TKI-sensitive and resistant models, including those with EGFR T790M and C797S mutations. We further uncover that EGFR homo- or heterodimerization with other ERBB family members, as well as the EGFR L747S mutation, confers resistance to JBJ-09-063, but not to ATP-competitive EGFR TKIs. Overall, our studies highlight the potential clinical utility of JBJ-09-063 as a single agent or in combination with EGFR TKIs to define more effective strategies to treat EGFR-mutant lung cancer.
Silent mutations reveal therapeutic vulnerability in RAS Q61 cancers
Yoshihisa Kobayashi, Chhayheng Chhoeu, Jiaqi Li, Kristin S. Price, Lesli A. Kiedrowski, Jamie L. Hutchins, Aaron I. Hardin, Zihan Wei, Fangxin Hong, Magda Bahcall, Prafulla C. Gokhale & Pasi A. Jänne.

Cancers with KRAS Q61X mutations rely on a secondary silent mutation G60G for survival and can be selectively treated with a mutant-specific antisense oligonucleotide.
Abstract:
RAS family members are the most frequently mutated oncogenes in human cancers. Although KRAS(G12C)-specific inhibitors show clinical activity in patients with cancer1,2,3, there are no direct inhibitors of NRAS, HRAS or non-G12C KRAS variants. Here we uncover the requirement of the silent KRASG60G mutation for cells to produce a functional KRAS(Q61K). In the absence of this G60G mutation in KRASQ61K, a cryptic splice donor site is formed, promoting alternative splicing and premature protein termination. A G60G silent mutation eliminates the splice donor site, yielding a functional KRAS(Q61K) variant. We detected a concordance of KRASQ61K and a G60G/A59A silent mutation in three independent pan-cancer cohorts. The region around RAS Q61 is enriched in exonic splicing enhancer (ESE) motifs and we designed mutant-specific oligonucleotides to interfere with ESE-mediated splicing, rendering the RAS(Q61) protein non-functional in a mutant-selective manner. The induction of aberrant splicing by antisense oligonucleotides demonstrated therapeutic effects in vitro and in vivo. By studying the splicing necessary for a functional KRAS(Q61K), we uncover a mutant-selective treatment strategy for RASQ61 cancer and expose a mutant-specific vulnerability, which could potentially be exploited for therapy in other genetic contexts.
Kurppa KJ, Liu Y, To C, Zhang T, Fen M, Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, Portell A, Lizotte PH, Ficarro SB, Li S, Chen T, Haikala HM, Wang H, Bahcall M, Gao Y, Shalhout S. et al.

Inhibition of YAP signaling can eliminate drug-resistant tumor cells following EGFR TKI treatment.
Abstract:
Eradicating tumor dormancy that develops following epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) treatment of EGFR-mutant non-small cell lung cancer, is an attractive therapeutic strategy but the mechanisms governing this process are poorly understood. Blockade of ERK1/2 reactivation following EGFR TKI treatment by combined EGFR/MEK inhibition uncovers cells that survive by entering a senescence-like dormant state characterized by high YAP/TEAD activity. YAP/TEAD engage the epithelial-to-mesenchymal transition transcription factor SLUG to directly repress pro-apoptotic BMF, limiting drug-induced apoptosis. Pharmacological co-inhibition of YAP and TEAD, or genetic deletion of YAP1, all deplete dormant cells by enhancing EGFR/MEK inhibition-induced apoptosis. Enhancing the initial efficacy of targeted therapies could ultimately lead to prolonged treatment responses in cancer patients.
Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor
To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, et al.

Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor.
Abstract:
Allosteric kinase inhibitors offer a potentially complementary therapeutic strategy to ATP-competitive kinase inhibitors due to their distinct sites of target binding. In this study, we identify and study a mutant-selective EGFR allosteric inhibitor, JBJ-04-125-02, which as a single agent can inhibit cell proliferation and EGFRL858R/T790M/C797S signaling in vitro and in vivo. However, increased EGFR dimer formation limits treatment efficacy and leads to drug resistance. Remarkably, osimertinib, an ATP-competitive covalent EGFR inhibitor, uniquely and significantly enhances the binding of JBJ-04-125-02 for mutant EGFR. The combination of osimertinib and JBJ-04-125-02 results in an increase in apoptosis, a more effective inhibition of cellular growth, and an increased efficacy in vitro and in vivo compared with either single agent alone. Collectively, our findings suggest that the combination of a covalent mutant–selective ATP-competitive inhibitor and an allosteric EGFR inhibitor may be an effective therapeutic approach for patients with EGFR-mutant lung cancer.
KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS
Ambrogio C, Kohler J, Zhou ZW, Wang H, Paranal R, Li J, et al..

KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS.
Abstract:
The mechanism by which the wild-type KRAS allele imparts a growth inhibitory effect to oncogenic KRAS in various cancers, including lung adenocarcinoma (LUAD), is poorly understood. Here, using a genetically inducible model of KRAS loss of heterozygosity (LOH), we show that KRAS dimerization mediates wild-type KRAS-dependent fitness of human and murine KRAS mutant LUAD tumor cells and underlies resistance to MEK inhibition. These effects are abrogated when wild-type KRAS is replaced by KRASD154Q, a mutant that disrupts dimerization at the a4-a5 KRAS dimer interface without changing other fundamental biochemical properties of KRAS, both in vitro and in vivo. Moreover, dimerization has a critical role in the oncogenic activity of mutant KRAS. Our studies provide mechanistic and biological insights into the role of KRAS dimerization and highlight a role for disruption of dimerization as a therapeutic strategy for KRAS mutant cancers.
Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer
Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, et al..

METD1228V causes resistance to type I MET inhibitors, whereas sensitivity to type II MET inhibitors is maintained.
Abstract:
Amplified and/or mutated MET can act as both a primary oncogenic driver and as a promoter of tyrosine kinase inhibitor (TKI) resistance in non–small cell lung cancer (NSCLC). However, the landscape of MET-specific targeting agents remains underdeveloped, and understanding of mechanisms of resistance to MET TKIs is limited. Here, we present a case of a patient with lung adenocarcinoma harboring both a mutation in EGFR and an amplification of MET, who after progression on erlotinib responded dramatically to combined MET and EGFR inhibition with savolitinib and osimertinib. When resistance developed to this combination, a new MET kinase domain mutation, D1228V, was detected. Our in vitro findings demonstrate that METD1228V induces resistance to type I MET TKIs through impaired drug binding, while sensitivity to type II MET TKIs is maintained. Based on these findings, the patient was treated with erlotinib combined with cabozantinib, a type II MET inhibitor, and exhibited a response.
EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M.

Positions of missense mutations G719S and L858R and the Del-1 deletion in the three-dimensional structure of the EGFR kinase domain.
Abstract:
Receptor tyrosine kinase genes were sequenced in non-small cell lung cancer (NSCLC) and matched normal tissue. Somatic mutations of the epidermal growth factor receptor gene EGFR were found in 15 of 58 unselected tumors from Japan and 1 of 61 from the United States. Treatment with the EGFR kinase inhibitor gefitinib (Iressa) causes tumor regression in some patients with NSCLC, more frequently in Japan. EGFR mutations were found in additional lung cancer samples from US patients who responded to gefitinib therapy and in a lung adenocarcinoma cell line that was hypersensitive to growth inhibition by gefitinib, but not in gefitinib-insensitive tumors or cell lines. These results suggest that EGFR mutations may predict sensitivity to gefitinib.
MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA.

HCC827 GR cells are resistant to gefitinib in vitro and show MET amplification.
Abstract:
The epidermal growth factor receptor (EGFR) kinase inhibitors gefitinib and erlotinib are effective treatments for lung cancers with EGFR activating mutations, but these tumors invariably develop drug resistance. Here, we describe a gefitinib-sensitive lung cancer cell line that developed resistance to gefitinib as a result of focal amplification of the MET proto-oncogene. Inhibition of MET signaling in these cells restored their sensitivity to gefitinib. MET amplification was detected in 4 of 18 (22%) lung cancer specimens that had developed resistance to gefitinib or erlotinib. We find that amplification of MET causes gefitinib resistance by driving ERBB3 (HER3)-dependent activation of PI3K, a pathway thought to be specific to EGFR/ERBB family receptors. Thus, we propose that MET amplification may promote drug resistance in other ERBB-driven cancers as well.
Novel mutant-selective EGFR kinase inhibitors against EGFR T790M
Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Jänne PA.

Crystal Structure of WZ4002 bound to EGFR T790MA.
Abstract:
The clinical efficacy of epidermal growth factor receptor (EGFR) kinase inhibitors in EGFR-mutant non-small-cell lung cancer (NSCLC) is limited by the development of drug-resistance mutations, including the gatekeeper T790M mutation. Strategies targeting EGFR T790M with irreversible inhibitors have had limited success and are associated with toxicity due to concurrent inhibition of wild-type EGFR. All current EGFR inhibitors possess a structurally related quinazoline-based core scaffold and were identified as ATP-competitive inhibitors of wild-type EGFR. Here we identify a covalent pyrimidine EGFR inhibitor by screening an irreversible kinase inhibitor library specifically against EGFR T790M. These agents are 30- to 100-fold more potent against EGFR T790M, and up to 100-fold less potent against wild-type EGFR, than quinazoline-based EGFR inhibitors in vitro. They are also effective in murine models of lung cancer driven by EGFR T790M. Co-crystallization studies reveal a structural basis for the increased potency and mutant selectivity of these agents. These mutant-selective irreversible EGFR kinase inhibitors may be clinically more effective and better tolerated than quinazoline-based inhibitors. Our findings demonstrate that functional pharmacological screens against clinically important mutant kinases represent a powerful strategy to identify new classes of mutant-selective kinase inhibitors.
Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer
Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, Jänne PA

Cotargeting EGFR and MEK prolongs effective treatment duration in EGFRL858R/T790M genetically engineered mice.
Abstract:
Irreversible pyrimidine-based EGFR inhibitors, including WZ4002, selectively inhibit both EGFR-activating and EGFR inhibitor-resistant T790M mutations more potently than wild-type EGFR. Although this class of mutant-selective EGFR inhibitors is effective clinically in lung cancer patients harboring EGFR (T790M), prior preclinical studies demonstrate that acquired resistance can occur through genomic alterations that activate ERK1/2 signaling. Here, we find that ERK1/2 reactivation occurs rapidly following WZ4002 treatment. Concomitant inhibition of ERK1/2 by the MEK inhibitor trametinib prevents ERK1/2 reactivation, enhances WZ4002-induced apoptosis, and inhibits the emergence of resistance in WZ4002-sensitive models known to acquire resistance via both T790M-dependent and T790M-independent mechanisms. Resistance to WZ4002 in combination with trametinib eventually emerges due to AKT/mTOR reactivation. These data suggest that initial co-targeting of EGFR and MEK could significantly impede the development of acquired resistance in EGFR-mutant lung cancer.
Significance:
Patients with EGFR-mutant lung cancer develop acquired resistance to EGFR and mutant-selective EGFR tyrosine kinase inhibitors. Here, we show that co-targeting EGFR and MEK can prevent the emergence of a broad variety of drug resistance mechanisms in vitro and in vivo and may be a superior therapeutic regimen for these patients.
Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M
Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Jänne PA, Oxnard GR.

Acquired resistance to AZD9291 mediated by acquired EGFR C797S mutation.
Abstract:
Here we studied cell-free plasma DNA (cfDNA) collected from subjects with advanced lung cancer whose tumors had developed resistance to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) AZD9291. We first performed next-generation sequencing of cfDNA from seven subjects and detected an acquired EGFR C797S mutation in one; expression of this mutant EGFR construct in a cell line rendered it resistant to AZD9291. We then performed droplet digital PCR on serial cfDNA specimens collected from 15 AZD9291-treated subjects. All were positive for the T790M mutation before treatment, but upon developing AZD9291 resistance, three molecular subtypes emerged: six cases acquired the C797S mutation, five cases maintained the T790M mutation but did not acquire the C797S mutation, and four cases lost the T790M mutation despite the presence of the underlying EGFR activating mutation. Our findings provide insight into the diversity of mechanisms through which tumors acquire resistance to AZD9291 and highlight the need for therapies that are able to overcome resistance mediated by the EGFR C797S mutation.